
Model of Organ Fibrosis
Mouse Model for the Study of Multi-Organ Fibrosis
Novel Inducible Fibroblast Specific Spag17 Knockout Mouse
Fibrotic disease is often the result of persistent myofibroblast activation and is a hallmark of systemic sclerosis. Systemic sclerosis is unique in that the fibrosis targets the skin, hearts, lungs, and muscle tissue, making this disease debilitating and deadly. Unfortunately, with few models to study systemic sclerosis and other fibrotic diseases, treatments for patients afflicted with these diseases are not common. Researchers at Virginia Commonwealth University (VCU) have identified and developed a novel mouse model that mimics the fibrotic phenotype observed in patients afflicted with multi-organ fibrotic diseases.
The technology
The novel mouse model created is an inducible fibroblast specific Spag17 knockout. Cells deficient in Spag17 also express an EGFP fluorophore for in vivo tracking. Investigators identified the Spag17 gene as one that is differentially regulated in fibrotic diseases such as scleroderma. Knockout Spag17 mice show significant fibrosis of their skin, heart, lungs, kidneys, and skeletal tissue between 4-6 months of age as shown in the figure below. Evaluation shows that these mice also have greater collagen deposition, enhanced fibroblast to myofibroblast transition, and changes in morphogen activity. The generated mouse model mimics the pathophysiology of systemic sclerosis and will present useful avenues to study multi-organ fibrosis as a result of primary cilia disfunction leading to fibroblast to myofibroblast transition.
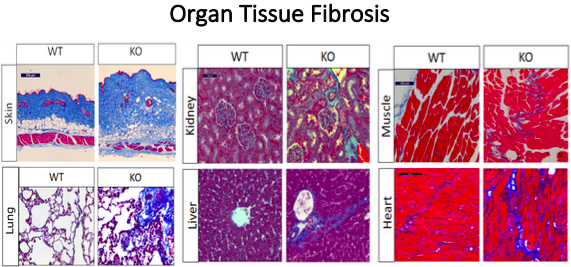
Figure 1. Multi-organ fibrosis is one of the phenotypes developed after conditional deletion of Spag17. Shown above are representative histological sections from skin, lung, kidney, liver, muscle and heart tissue stained by Masson’s Trichrome stain as a fibrotic marker. Increased collagen deposition (blue) is observed in Spag17 knockout mice compared to wild-type controls.